病毒基因组信息可用于研究病毒的致病机理和进化,以及协助疫苗开发和疫苗耐药性研究。现在可以通过表征来自不同宿主甚至单个个体的病毒基因组来详细探索病毒突变。例如,在过去两年中发现了数千种 SARS-CoV-2 变体,其中包括世界卫生组织在本书出版时确定的几种“令人担忧的变体” 。
由于读取测序技术以及序列组装和分析软件的进步,病毒基因组菌株的识别和特性分析现在已成为一项常规任务。目前已发布数千个病毒基因组,并且每天都有更多病毒基因组被添加。
然而,尽管病毒基因组分析任务可能很常规,但许多研究人员发现它令人沮丧且耗时。DNASTAR 最近调查了 100 多名病毒学家和基因组学研究人员,了解他们在识别和表征病毒样本方面面临的挑战。他们指出,大多数组装和分析软件都存在一些缺陷,这是轻松执行此工作流程的重大障碍。

识别和表征病毒株的一般程序是什么?
识别和鉴定病毒株主要涉及四个步骤。
第一步甚至在您将样本送去测序之前就已完成。

步骤 1:确定是否将实验序列读取组装到参考文献中或从头组装
在决定测序技术(步骤 2)之前,重要的是要知道您是从头组装序列还是根据参考序列组装序列。
如果您研究的是新型病毒,或者您的病毒突变率高或变化范围大(如分段重配),则无法获得紧密匹配的参考基因组。对于这些病毒,别无选择,只能进行从头组装。
对于更稳定的病毒基因组,突变率相对较慢,可以使用来自相同(或密切相关)菌株的基因组参考序列来指导测序读数的比对。如果您感兴趣的病毒有参考序列,我们强烈建议您使用它。模板化组装有助于将重叠群排序为更大的支架,为您提供更完整的基因组序列,并允许更快、更准确地组装。这些组装非常适合监测 SARS-CoV-2 等病毒株的进化。下游分析通常侧重于识别定义病毒株或影响病毒蛋白的变异。
第 2 步:选择测序技术并提交样本
如果您要进行参考引导组装,Sanger 或 Illumina 测序技术非常适合病毒基因组分析。这些技术产生的读取比一些较新的技术更短,但仍然非常准确。
如果您要进行从头组装,您选择的测序技术将取决于您正在研究的病毒基因组的长度。使用 Sanger 和 Illumina 2X300bp MiSeq 读段可以对长度高达 10Kbp 的病毒基因组进行出色的从头组装。但是,使用长读段最好能够对较长和/或更复杂的基因组(如 SARS-COV-2 (30Kbp) 或流感病毒(分段基因组))进行准确的从头组装。
长读序列数据比早期的测序类型具有许多优势。在本书出版时,主要有三种长读测序技术:Oxford Nanopore Technologies (ONT/Nanopore)、Pacific Biosciences (PacBio) CLR 和 PacBio HiFi。
尤其是 PacBio HiFi 测序,它能产生长而准确的(>99.9%)序列读取,非常适合参考引导和从头组装。PacBio HiFi 读取是通过 PacBio 的 Sequel HiFi 测序平台产生的,使用一种称为“循环共识测序”(CSS)的混合短读和长读测序器模式。PacBio HiFi 读取比使用其他两种长读测序技术产生的长读更短,但更准确。由于这种高水平的准确性,使用 PacBio HiFi 技术测序的病毒基因组数据通常会组装成单个重叠群。
决定了测序技术后,请在内部对实验样本进行测序或将其提交给核心机构。如果您选择了长读技术,请注意,大多数小型测序机构无法使用长读测序平台。要获得这些格式的读段,您可能需要将样本发送到大型外部机构。
根据设施情况,您将得到原始序列读数或草图基因组。
如果您要从原始序列读取开始,请继续执行步骤 3,在此您将把这些读取组装成草图基因组。如果您已经有草图基因组,请转到步骤 4,在此您将分析进化关系并找到感兴趣的序列和变体。
![]()
步骤 3:将原始读段组装成一组草图基因组
如果您从原始测序读段开始,则需要使用软件将它们组装成一组草图基因组。几十年前,这一步可能需要长达一个小时。如今,如果您使用涵盖整个病毒基因组的长读段,现代软件可以在短短几秒钟内完成组装。
对于从头组装,您不需要任何其他东西,只需要组装软件和序列读取文件(通常为 FASTQ 格式)。组装软件将生成一个或多个代表病毒基因组草图的重叠群。我们建议选择允许您可视化和编辑这些重叠群的软件,以生成最准确的基因组草图。然后可以将此基因组草图导出为 FASTA 格式,并在步骤 4 中与其他病毒株进行比较。
如果您有一个密切相关的参考基因组,您可以使用它来指导原始测序数据的组装。美国国家生物技术信息中心 (NCBI) 是一个优秀的参考基因组来源,可以免费下载。点击此处访问 NCBI 的核苷酸参考基因组搜索页面。然后可以检测测序菌株和参考基因组之间的变异,并将其写入变异报告或 VCF 文件,以便在第 4 步进行进一步分析。
步骤 4:将实验草图基因组相互比较和/或与参考序列进行比较
如果您的实验基因组草图是使用参考文献组装的,那么您可能会对查找重要变异感兴趣。寻找支持过滤的软件,以便您可以将发现的变异限制为感兴趣的 SNP 和插入/缺失。理想的软件应支持变异调用的常见文件格式,如 VCF,还应允许您使用特定过滤标准轻松比较多个样本中的变异。
多序列比对可用于将草图基因组相互比较或与相关参考序列进行比较。在比对大量病毒基因组时,有许多可用的算法。然而,MAFFT7 因其准确性和高容量而被认为在大量病毒基因组中更胜一筹。
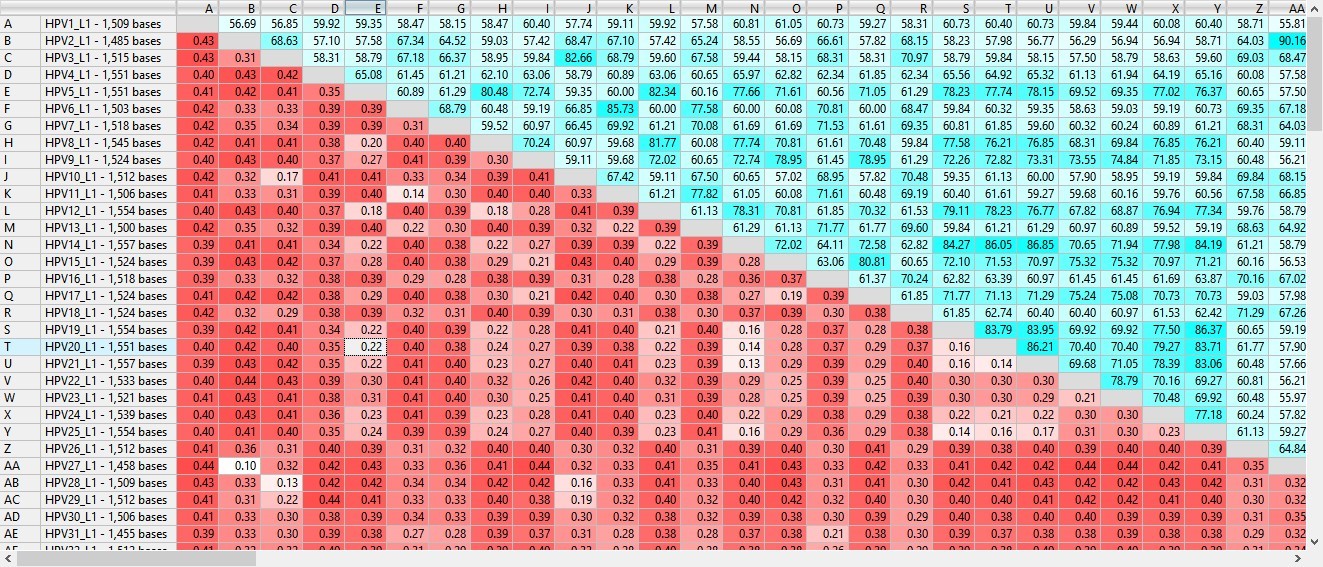
图 1. MegAlign Pro距离表的部分,包含与 MAFFT 比对的 10,000 个病毒样本。
经过多重序列比对后,结果可以以距离表(图 1)或系统发育树的形式查看。如果您想要了解两种菌株之间的差异,某些软件可以让您使用针对成对比较优化的算法仅比对这两个序列。
请参阅下一页的方框,其中有一个示例,了解如何使用多序列比对来识别实验对象中的 SARS-CoV-2 病毒株。
示例:使用系统发育树识别实验病毒样本的菌株
此示例取自使用 MegAlign Pro 多序列组装软件的 DNASTAR教程。
首先,将来自未知 SARS-CoV-2 毒株的受试者的实验病毒序列(SRR13380669_ NC_045512.2)上传到多序列比对应用程序。还上传了 NCBI 中四种已知 SARS-CoV-2 毒株的参考序列(见表)。
接下来,选择MAFFT算法,使用所有上传的序列进行多序列比对。
根据结果比对结果自动生成系统发育树(图 2)。下图中,实验样本SRR13380669_NC_045512.2显示为具有绿色背景的进化枝的一部分,即 B-1-351,或 SARS-CoV-2 的“Beta”菌株。
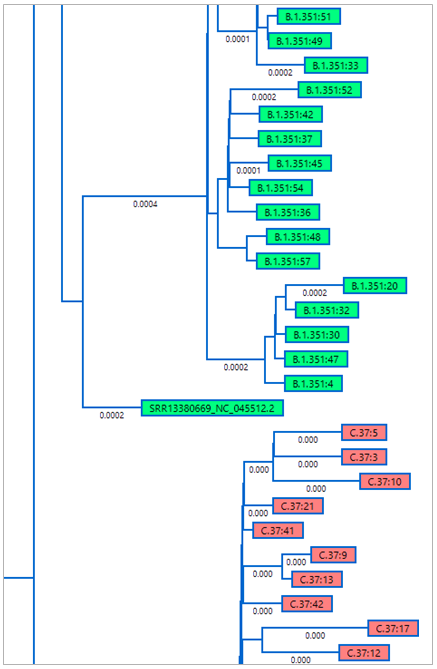
图 2. MegAlign Pro 系统发育树的一部分,显示了实验样本在“Beta”菌株进化枝中的位置。
为了验证,比对距离表自动按与实验样本相关性从高到低进行排序(图3)。相关性最高的样本全部来自“Beta”菌株,进一步证实了SRR13380669_NC-045512.2确实来自该菌株。
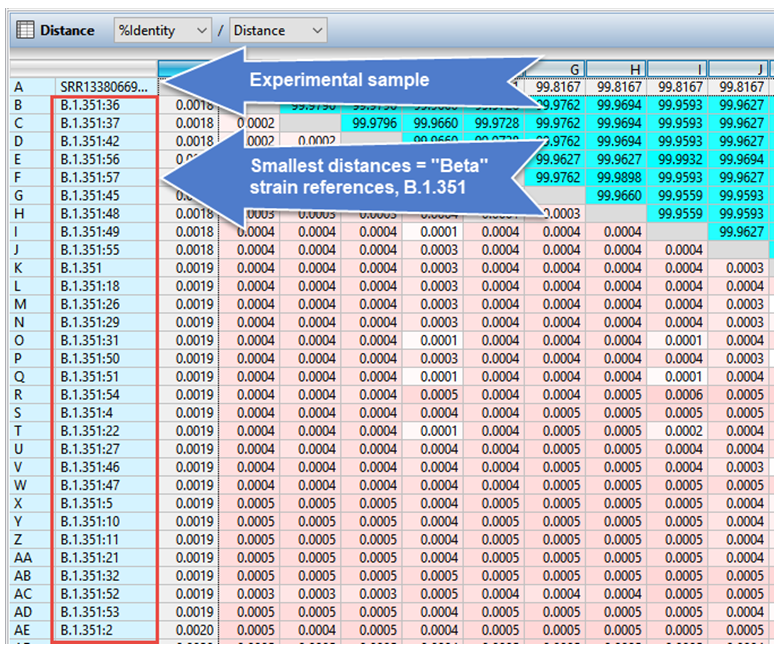
图 3. MegAlign Pro 距离表的一部分确认了实验样本在“Beta”菌株进化枝中的位置。
