第三方比较:Lasergene Genomics 与CLC Genomics Workbench
我们认为Lasergene Genomics中的从头转录组工作流程在同类产品中处于领先地位,并且 2018 年的这项研究也认为SeqMan NGen在多个领域的工作流程表现都优于 CLC Genomics Workbench。
下面,我们将详细阐述论文中提出的一些发现。
灵活的装配设置
研究作者报告称,SeqMan NGen “……允许用户在组装之前指定 rRNA 或其他输入污染物序列。此选项目前在 CLC GW 从头转录组工作流程中不可用。”
除了让您指定 rRNA 和其他污染物序列外,SeqMan NGen 向导还允许您移除特定的载体或接头序列(图 2)。或者,您也可以选择通过选中“移除通用接头”选项来执行全自动接头移除。
更少、更长的重叠群
在其他应用中, RNA-Seq 数据的从头组装可能会产生数千个代表表达转录本的未标记重叠群。对如此多未注释的重叠群进行有意义的下游分析几乎是不可能的。
然而,使用其专有的组装算法,SeqMan NGen 产生的重叠群比 CLC Genomics Workbench 更少、更长。研究作者指出,“Lasergene SMN Trace Evidence 共识调用算法平均产生更长的重叠群……与此同时,CLC GW 组装的重叠群数量是其九倍以上……”
SeqMan NGen 如何做到这一点?SeqMan NGen 会自动尝试将来自同一基因的重叠群分组,然后根据与从 NCBI 的 RefSeq 网站数据中提取的注释参考序列集合(“转录本注释数据库”)的最佳匹配来命名和注释它们。与 RefSeq 序列对齐和匹配的转录本片段总数提供了测序覆盖率。使用 SeqMan NGen 组装的许多数据集会产生大量可能是全长转录本的长转录本。
污染物序列报告
缺乏报告排除读取功能的软件可能会对读取进行过度采样,从而降低转录组组装的精度。相比之下,SeqMan NGen 会报告哪些读取被排除。比较研究发现,SeqMan NGen “……在其项目报告中明确定义了排除读取……”
下游分析能力
从头转录组组装之后,Lasergene Genomics 包中的其他应用程序允许不同类型的下游分析。
综合报告
想知道您是否看到了新的东西?在SeqMan Ultra中打开完成的组装,在两个高度可定制且可排序的报告中分别查看已知和新的转录本(图 11)。
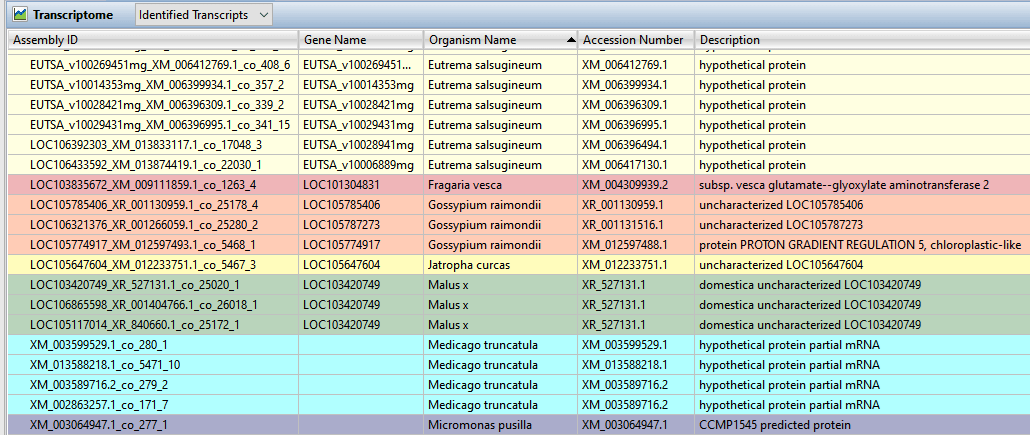
图 11 .SeqMan Ultra 的“已识别转录本”报告按生物体名称以颜色编码显示。
据研究作者称,SeqMan NGen “生成了注释和新的转录本列表。NCBI RefSeq 数据库用于从组装的转录本序列中获得大量已知或同源基因。”相比之下,“CLC GW 组装输出包含组装转录本和未组装序列读数的列表。”
要查看 DNASTAR 针对不同数据集比较已识别和组装的新转录本的基准,请参阅此博客文章。
顺便说一句,如果您好奇为什么软件找到的平均转录本长度通常比生物体 mRNA 的长度短,上面的博客文章也解释了这一现象。简而言之,读取长度对从头转录组组装有很大影响。Illumina 读取长度超过 150bp 通常会产生更长的组装转录本 - 最长可达全长 - 而读取长度小于 150bp 的转录本可能只有 mRNA 长度的一半。
集成热图和基因本体
您可以使用ArrayStar将转录组结果以热图的形式查看(图 12),并对转录本进行基因表达分析。
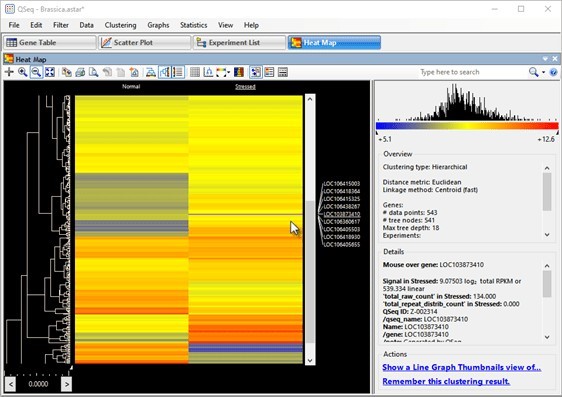
图 12.芸苔属植物中两种组织类型的差异表达,如 ArrayStar 热图所示。
您还可以使用 ArrayStar 探索基因本体。据该论文的作者称,“基因本体 (GO) 分析提供了基因的功能描述以及基因之间的现有关系或功能节点。” SeqMan NGen “有一个集成工具可以执行 GO 分析,但没有 CLC Genomics Workbench。”
