使用 Lasergene Genomics 简化工作流程
Lasergene Genomics软件包中的三个应用程序- SeqMan NGen、SeqMan Ultra和ArrayStar - 共同作用,作为从头转录组组装和下游分析的集成流程。
去除污染物并组装和注释转录本
SeqMan NGen 是一款一体化工具,用于去除接头和其他污染物、组装转录本,然后使用组特定注释自动对其进行注释。此工具仅需 1-2 分钟的手动操作时间即可完成组装,并涵盖了本电子书上一节中的“软件步骤 1-3”。
要设置组装,启动 SeqMan NGen 并选择“ De novo转录组”工作流程(图 3)。

图 3.在 SeqMan NGen 向导中选择工作流程。
之后,按照提示指定自动删除最常见的适配器,包括 Illumina 通用适配器,以及您指定的任何 rRNA 污染序列或载体(图 2和4)。

图 4 . SeqMan NGen 可让您通过勾选复选框并添加一个或多个污染物文件来筛选特定的 rRNA 污染物。
然后您上传您的从头转录组读数。
在设置过程中,请务必选中复选框(图 5)以使用 DNASTAR 的专有转录本注释数据库自动注释转录本。

图 5.在 SeqMan NGen 向导中选择所需的转录本注释数据库。
单击下载数据库按钮并从相关分类群中选择一个 mRNA 数据库或选择整个 RefSeq 数据库。这些数据库是参考 mRNA 集,包括从NCBI 的 RefSeq 网站数据中提取的序列数据和转录注释。由于您是从广泛的生物群体而不是单一物种中进行选择,因此这些注释可用于促进来自十几个生物群体的模型和非模型生物的组装和注释。(注意:如果您的实验室无法访问互联网,您可以选择使用 RefSeq 的本地副本作为注释数据库)。
项目设置完成后,您可以选择在本地计算机或云端进行组装。SeqMan NGen 的专利聚类和组装算法可以在标准台式计算机上组装多达 5 亿个读数,无需超级计算集群。
在组装过程中,SeqMan NGen 会自动尝试将来自同一基因的重叠群分组,然后根据与注释参考序列集合的最佳匹配来命名和注释它们。使用多种组装算法来优化结果。
分析记录
组装完成后,双击 SeqMan NGen 输出的 .transcriptome 文件,在 SeqMan Ultra 中打开结果。结果显示为两个交互式表格:一个用于识别的转录本(如果您选择使用 TAD),另一个用于新转录本(图 6)。

图 6 . SeqMan Ultra 中的菜单可让您指定要显示的转录本表。
您可以添加或删除包含数十种信息类型的列,从转录本长度到与该转录本相关的基因名称。SeqMan Ultra 的综合用户指南包含每个可用列的描述。单击相应的列标题,按任何数据类型的字母数字顺序对表格进行排序。系统会自动应用颜色,以便更容易看到具有相同质量的转录本分组(图 7)。

图 7.SeqMan Ultra 转录组视图中已识别的转录本。
请注意,如果您稍后再次进行分析,新转录本和已识别转录本的数量将发生变化。这是因为转录组注释数据库 (TAD) 会随着新转录本的添加而增长。这意味着特定生物体的“已识别”转录本的数量将增加,“新”转录本的数量将减少。
SeqMan NGen从头转录组工作流程的基准
DNASTAR 的从头转录组组装自动注释方法可生成许多已识别的转录本,甚至许多非模式生物也是如此。表 1显示了此工作流程的基准。使用 Lasergene Genomics 版本 17.3 所需的组装时间约为使用 SeqMan NGen 先前版本组装这些组所需的时间的一半。
表 1.使用 Lasergene Genomics 17.3 的SeqMan NGen从头转录组组装工作流程的基准。
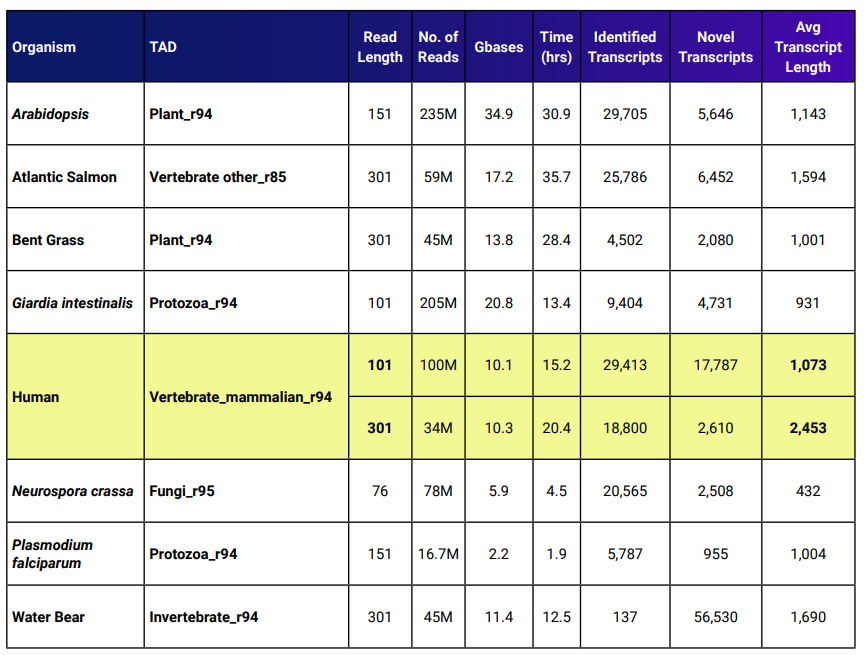
黄色行显示了人类数据的两个基准。一个数据集的平均读取长度是另一个数据集的 2.98 倍。较长读取的数据集的平均转录本长度是较短读取的平均转录本长度的 2.29 倍。
分析基因表达
如果您使用转录组数据来识别差异基因表达,筛选大量数据可能会很麻烦。如果您研究的是非模式生物,那么缺乏可用的参考序列会使使用典型软件解决方案的工作流程变得极其困难甚至不可能。
但有了 Lasergene Genomics,无论你研究的是哪种生物,基因表达分析都变得非常简单。在完成上述从头转录组组装后,只需再进行两个步骤。
首先,使用 SeqMan NGen 遵循模板化的 RNA-Seq 组装工作流程(图 8)。
图 8 . 模板化的 RNA-Seq 工作流程在 SeqMan NGen 的“工作流程”屏幕上的位置。
当提示添加样本序列时,上传您用于创建从头转录组组装的相同数据集。当提示添加参考序列时,单击添加文件夹并上传从头转录组组装的 .fasta 输出。您可以选择使用完全注释的已识别转录本、新转录本或两者(图 9)。
图 9.指定在 RNA-Seq 组装中使用哪些转录本作为参考。
如果您在 SeqMan Ultra 进行分析时导出了转录本子集,您也可以通过单击“添加”并添加导出的 .fas 文件来使用它们。
其次,在 ArrayStar 中打开结果,量化本项目中使用的转录本的相对丰度并分析基因表达。按照上述步骤操作意味着与转录本相关的基因名称可以自动显示在 ArrayStar 中。ArrayStar 的高级过滤工具、强大的统计分析和丰富的图形视图可让您轻松分离感兴趣的基因组并确定其生物学意义和本体论。
在下面的例子中,非模式生物的转录组数据用于识别实验中上调的基因。首先,基因表按其 log2 倍数变化列排序,以便“诱导”实验中上调的基因出现在表格顶部。然后勾选 11 个上调最多的基因旁边的框,使它们在相关散点图中显示为白点(图 10)。这些基因可以保存为一组以供进一步分析。
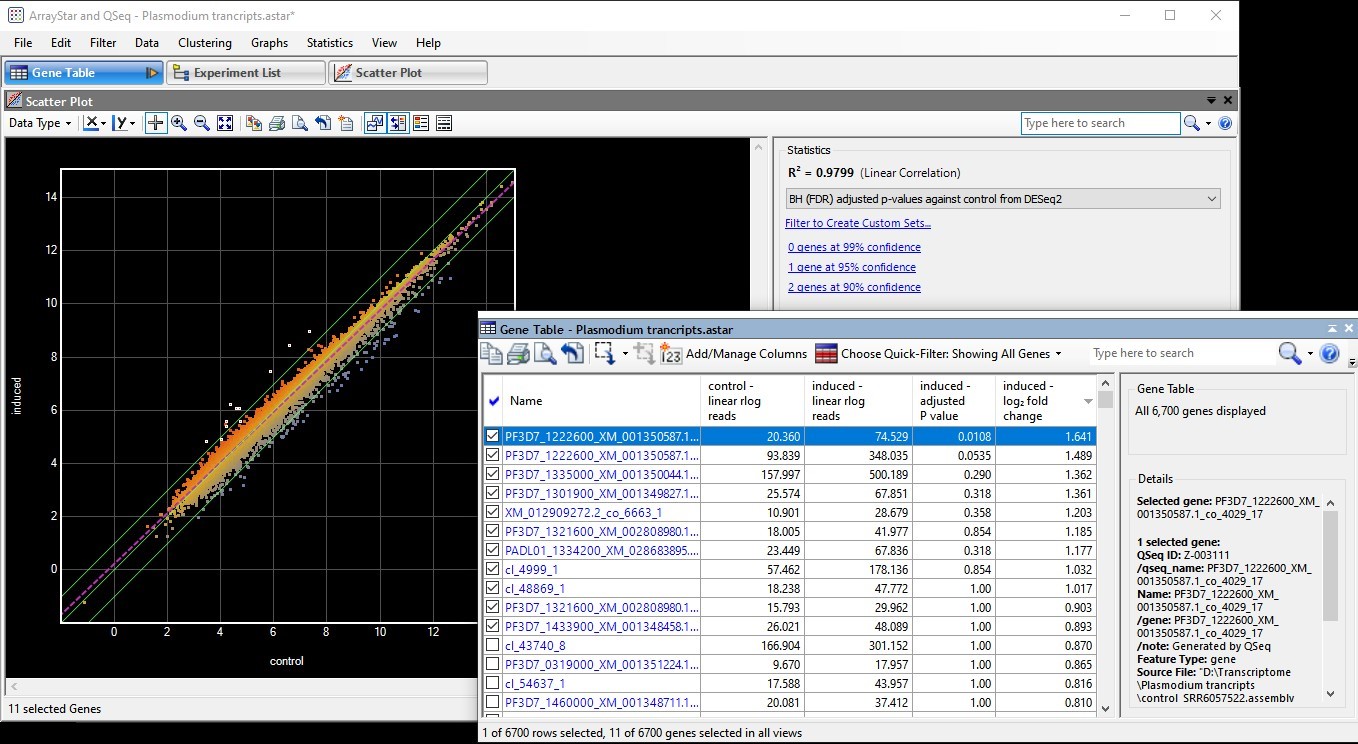
图 10 . 在“诱导”实验中选取上调最多的基因(白色)后的 ArrayStar 散点图。
