本章将讨论 AlphaFold 2 和 AlphaFold-Multimer 的优势以及利用其开源版本的一些障碍。
AlphaFold 2 和 AlphaFold Multimer 有什么特别之处?
AlphaFold 2 由 Alphabet Inc.(谷歌)子公司 DeepMind 开发。这种独特的算法使用人工智能/机器学习来计算蛋白质的可能 3D 结构。

在过去的八次 CASP 实验中,根据“常规”目标的综合组 z 分数,仅有三组在比赛中获得第一名。(表 1)。
| 事件 | 年 | 优胜者 |
| CASP7 | 2006 | 张 |
| CASP8 | 2008 | 张 |
| CASP9 | 2010 | 张 |
| CASP10 | 2012 | 张 |
| CASP11 | 2014 | 张 |
| CASP12 | 2016 | 贝克(罗塞塔小组) |
| CASP13 | 2018 | A7D |
| CASP14 | 2020 | AlphaFold 2 |
| CASP15 | 2022 | Yang-Server(AlphaFold 2 未参赛) |
表 1. 2006 年至今每个 CASP 实验中排名最高的算法,基于常规目标的组合组 z 分数。使用颜色表示来自同一组的算法。AlphaFold 2 是“A7D”算法的更新迭代。“Zhang”是 DNASTAR NovaFold 使用的 I-TASSER 算法的另一个名称,而“Yang-Server”是来自同一组的不同算法。
I-TASSER(在 CASP 活动中称为 Zhang 或 Yang-Server)比任何其他算法都赢得了更多的 CASP 实验,并构成了 DNASTAR 的NovaFold应用程序的基础。
2020 年 CASP14 的结果带来了转折点。AlphaFold 2 作为首次参加 CASP 的选手击败了其他 145 种算法,在蛋白质研究界引起了轰动。AlphaFold 2 不仅赢得了这场全球实验,而且其准确率比最接近的竞争对手 Baker(Rosetta Group)算法(赢得 CASP13)高出 2.65 倍(图 1)。
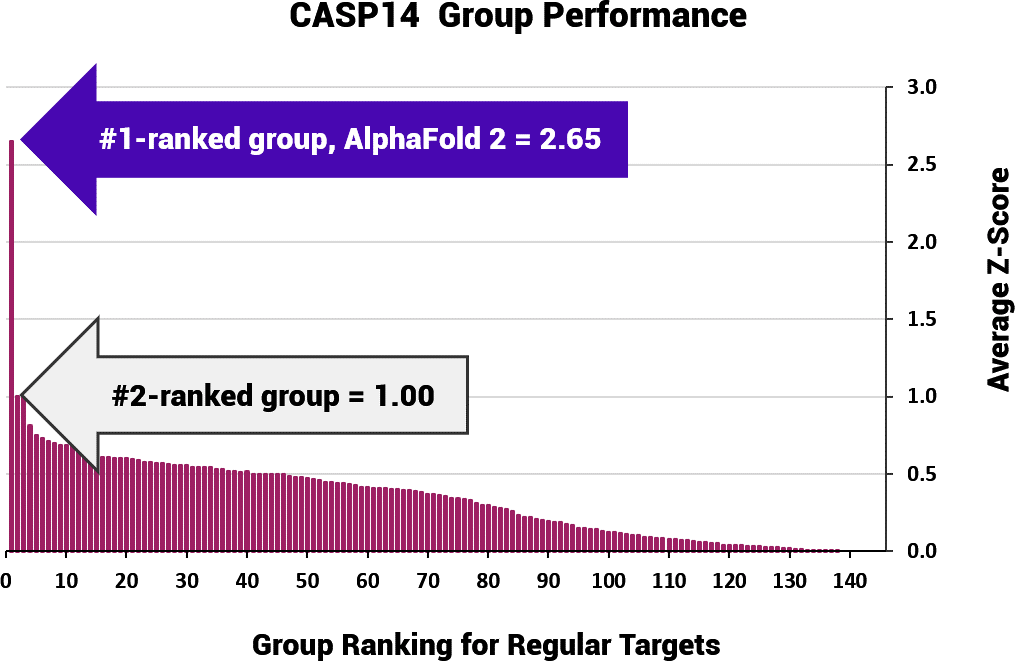
图 1. 在 CASP14 挑战赛中,AlphaFold 2 在 92 个“常规”目标上取得了 2.65 的平均 z 分数*。最接近的竞争对手在相同数量的目标上只取得了 1.00(图表由此表创建)。
* 调整后的 z 分数,本质上是高于全套模型平均值的标准差 (SD) 数,是在最近的 CASP 实验中使用 Croll TI、Sammito MD 等人的协议计算得出的。CASP13 中基于模板的建模评估,蛋白质,第 87 卷,第 12 期,第 1113-1127 页。
根据 CASP 结果和全球真实用户的经验,AlphaFold 2 是目前最准确、速度最快的单链蛋白质预测算法,可在几小时甚至更短的时间内完成大多数结构预测。AlphaFold 2 还可以预测非常具有挑战性的蛋白质的 3D 结构,例如:
- 膜结合蛋白
- 融合蛋白
- 胞质结构域 (CD)
- 细胞外区域
- G 蛋白偶联受体 (GPCR)
该算法还可以模拟多个域及其与接头的相互作用,也称为多域蛋白质结构预测。
AlphaFold-Multimer 是 AlphaFold 2 算法的扩展,由 DeepMind 于 2021 年推出。与 AlphaFold 2 相比,AlphaFold-Multimer 使用了两个额外的指标来评估预测准确性。要了解有关这些的更多信息,请参阅此EMBL-EBI 训练页面。
AlphaFold-Multimer 尚未以自己的名义进入 CASP。然而,在 CASP15 (2022) 的多聚体目标类别中,表现最好的三名算法(这是最近计算结果的实验)都使用该算法作为其方法的基础元素。因此,AlphaFold-Multimer 被认为是预测蛋白质-蛋白质复合物四级结构的领先算法。
要了解有关 AlphaFold2 算法的更多信息,请参阅 Jumper, J、Evans, R、Pritzel, A 等人。使用 AlphaFold 进行高精度蛋白质结构预测。Nature 596, 583–589 (2021)。要了解有关 AlphaFold-Multimer 的更多信息,请参阅 Evans R、O'Neill, M 等人。(2021)。使用 AlphaFold-Multimer 进行蛋白质复合物预测。bioRxiv 2021(预印本)
免费访问 AlphaFold 模型
EMBL-EBI 的 AlphaFold 数据库有 2 亿个结构预测,可供免费下载。绝对值得检查一下数据库中是否有您感兴趣的蛋白质的预测模型。
尽管如此,仍有大量蛋白质尚未被预测并放入数据库中,也不会被放入数据库中。这是因为 EMBL-EBI 数据库仅包含天然存在的蛋白质,而且只包含其中的一部分。该数据库不包括定制融合蛋白质、合成蛋白质和天然存在的蛋白质的突变。此外,该数据库仅包含每种蛋白质的一个预测结构,不包含替代剪接版本或异构体。
由于这些限制,如果您想了解自己感兴趣的蛋白质,您可能需要设置并运行一种新的预测。下一节将介绍此工作流程。

使用开源 AlphaFold 2 和 AlphaFold-Multimer 预测新结构
AlphaFold 2 和 AlphaFold-Multimer 可以免费下载和使用,但它们具有上一章中描述的开源软件的许多固有缺点。与其他开源预测软件一样,它们通常需要 IT 团队的协助才能设置,并通过在命令行中输入复杂命令来运行(见表 2)。
此外,这些算法在本地机器上运行,而不是在云端,并且必须安装在性能极高、容量极高的 Linux 计算机上。例如,AlphaFold 2 及其相关模板库(对于每个预测的序列搜索和比对阶段至关重要)需要 2.5 TB 的磁盘空间。对于大多数蛋白质研究人员来说,购买昂贵的 Linux 机器仅用于单个应用程序是不切实际的。
最后要考虑的是,这些算法的开源版本不包含蛋白质结构查看器。因此,您需要安装和学习其他软件才能查看预测的蛋白质模型。
如果您已经拥有安装了 Docker 的 Linux 计算机,并且熟悉使用命令行脚本,那么还有另外两种可用的替代方案:
对于 AlphaFold 2,有一个在云端运行的AlphaFold Colab,所需的磁盘空间比上面描述的完整 AlphaFold 2 安装少得多。但是,此解决方案使用了简化版的 AlphaFold 2,其准确率低于完整版,并且不适用于多链蛋白质分子(多聚体)。
AlphaFold-Multimer 可通过ColabFold运行,它在 Google Colab 笔记本的限制范围内运行。但是,Google Colab 笔记本存在内存限制,这可能会给运行 AlphaFold-Multimer 算法带来挑战。
